
The concept of pseudoalignment was first introduced in Nicolas Bray’s PhD Thesis and is one of the core concepts behind the fast RNA-Seq quantification in kallisto.
A pseudoalignment of a read is simply a set of target sequences that the read is compatible with. Contrary to normal read alignment, where we specify where the read aligns and how, i.e. how the nucleotides in the read match up with the nucleotides in the target sequence. A more precise definition and discussion can be found in Lior Pachter’s blog post What is a read mapping.
The definition of pseudoalignment is crafted so that the output is a sufficient statistic for the basic RNA-Seq quantification model. As such, the focus is on sets of transcripts with counts rather than detailed alignments, and this makes pseudoalignments less intuitive than regular alignments, and harder to visualize.
Early on in the development of kallisto I started working on a way to report each pseudoalignment in a systematic way, namely to convert them to SAM records. This started only for debugging purposes, but we thought it was useful so we added it to the early releases. We called this approach pseudobam (even though the output was SAM, a text format which could be converted to BAM, a binary representation). In addition to reporting the set of targets the read aligns to we also included the position and orientation.
The issue was that SAM files are big and generating them took a long time, long by kallisto standards at least. Another annoyance was that they could only be computed using a single core, whereas regular kallisto can make use of multiple cores. Finally the output was only to transcriptome coordinates, since kallisto is by design annotation agnostic; it only requires transcript sequences and not the full genome or any knowledge of where those transcripts occur.
This has all changed now. In our latest release of kallisto (version 0.44.0) we output binary BAM files when kallisto is run with the pseudobam option. kallisto will make use of multiple cores when constructing the BAM file and additionally we report the posterior probability of each alignment in the BAM file. The posterior probability of an alignment is an estimate, based on the results of the EM algorithm that kallisto ran for quantification, of how likely the alignment was to have originated from that location1.
We also project the pseudoalignments down to a genome reference2, producing a BAM file in genomic coordinates, where the pseudoalignments which were originally computed w.r.t. transcripts have been turned into spliced alignments to the genome.
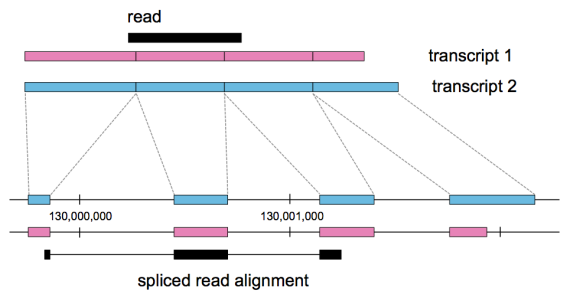
The BAM files that kallisto produces are automatically sorted by genome coordinates and indexed, so that they can be viewed immediately in a genome browser such as IGV. This circumvents the need for Samtools to sort and index the BAM file prior to visualization, in particular, kallisto now provides a simple and efficient reads to visualization for RNA-Seq analysis on windows machines.
When we were developing this we kept an eye on speed. Running kallisto and producing the BAM files took 24m11s using 8 cores on a file with 78M reads. When we tested this on unsorted reads generated by kallisto, using Samtools to sort and index required 13m45s using 8 threads. Although more thorough benchmarking is in order I think we can claim that we have the fastest reads-to-visualization pipeline currently available.
With your BAM files ready you can visualize your pseudoalignments, construct Sashimi plots and inspect the data interactively3.

IGV browser view of kallisto pseudoalignments of the LSP1 gene from RNA-Seq sample ERR1881406. The top panel shows a wiggle plot of coverage, with individual read pseudoalignments displayed below. Read pseudoalignments in the BAM file include the posterior probabilities in the ZW tag. The Sashimi plot shows the number of spliced reads identified by kallisto. The transcript structure is shown in the third panel.
When we’ve done differential expression studies using sleuth one of the things we do to verify the results is to plot the data for the top differentially expressed genes. We do this after having run a statistical test of every gene expressed. Similarly, we can now produce BAM files in genome coordinates to visulize the pseudoalignments and quality control them to ensure that our signal is not driven by artifacts or incomplete annotation.
Lior Pachter gave a keynote for the Genome Informatics where he showed some really exciting work on single cell RNA-Seq. Using this approach we could very quickly visualize the pseudoalignment and see where in the CD45 gene the 10X reads were mapping. The visualization of the data allowed us to pick up on the fact that the annotation of 3′ UTR is probably incomplete for most genes, something you don’t “see” when staring at a sea of reads.
Notes
- The concept of posterior probability of an alignment comes from eXpress and RSEM
- This genome projection option was also available as a script in TopHat and RSEM.
- The figures are from a poster I presented at Genome Informatics (full resolution poster)

 and a graph
and a graph 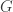 is there a path in the graph, whose sequence spells out something close (
is there a path in the graph, whose sequence spells out something close (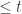 ) to the read
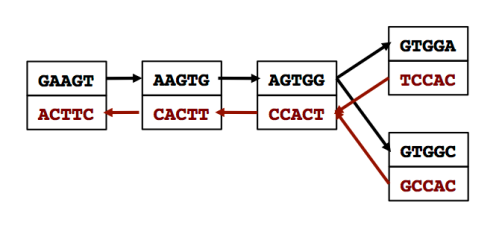
) to the read 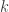 -mers in a de Bruijn Graph.
-mers in a de Bruijn Graph.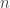 reads,
reads, 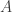 and BOB has a BAM file with
and BOB has a BAM file with 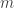 reads,
reads,  . They want to find out if
. They want to find out if  using minimal communication. This is equivalent to finding out if
using minimal communication. This is equivalent to finding out if  . Any fingerprint method can be turned into a protocol where Alice sends the fingerprint and Bob compares the fingerprints and answers yes or now. The general setting where Alice and Bob can talk interactively allows for more complicated methods. [edit: the direction was wrong, BAM are supposed to be subsets of FASTQ in a practical setting, for the lower bound you just reverse the roles of Alice and Bob, they won’t mind]
. Any fingerprint method can be turned into a protocol where Alice sends the fingerprint and Bob compares the fingerprints and answers yes or now. The general setting where Alice and Bob can talk interactively allows for more complicated methods. [edit: the direction was wrong, BAM are supposed to be subsets of FASTQ in a practical setting, for the lower bound you just reverse the roles of Alice and Bob, they won’t mind] , even if we allow randomness and some small probability of failure.
, even if we allow randomness and some small probability of failure.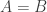 , which it called the EQ problem. The EQ problem has a lower bound of
, which it called the EQ problem. The EQ problem has a lower bound of 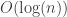 bit solution when we allow randomness, and this is pretty much what BamHash is doing.
bit solution when we allow randomness, and this is pretty much what BamHash is doing.

 such k-mers. So the number of k-mers is
such k-mers. So the number of k-mers is  , and for k odd we have
, and for k odd we have  distinct representatives, but for k even we have
distinct representatives, but for k even we have 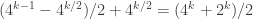 distinct representatives. [edit 4/30/15, thanks to Lior Pachter for noticing an arithmetic bug]
distinct representatives. [edit 4/30/15, thanks to Lior Pachter for noticing an arithmetic bug]