
tl;dr: yes
Earlier this year I attempted to review a paper that had an NP-completeness result for aligning reads to de Bruijn Graphs. I say attempted, because I received an invitation to review the paper, was late, did a detailed review anyway and then found out that the editor had removed me as a reviewer when I tried to submit it. I sent the review anyway, but I don’t know if the authors received it.
This paper just came out a few days ago: Read mapping on de Bruijn graphs
Papers come and papers go, I didn’t think more of it until recently on twitter
The language in this tweet is quite aggressive. Words like “bogus” and “certainly not NP-complete” do not (a) belong in a meaningful twitter conversation, (b) pinpoint any issue with the paper or (c) tell you why it’s wrong (after all the paper has a proof so it should be easy to tear that proof apart). I’m not going to go into point (a), that has been resolved on twitter.
What I want to get to are points (b) and (c). Before I do that, let’s look at the context. This paper has roughly two contributions, first the authors formulate a problem (I’ll get to what that means later) and show that this problem is NP-complete. Second the authors propose a quite reasonable heuristic that they show works well in practice and test on real data.
The formulation of the high level problem of aligning reads to a graph is as follows

Basically what this means is that the question is given a read 
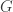
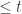
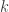
This seems like a reasonable formulation of the problem we really want to solve and quite close to the type of alignment problems we know how to deal with when aligning to a single reference sequence.
At this point it’s worthwhile to consider a different problem which plays a role in genomics (assembly), namely the Hamiltonian path problem. It is a problem which turns out to be NP-complete, yet it’s close relative, the Eulerian cycle problem is easily solved. This distinction is of value in guiding modeling choices for assembly and in algorithm design.
In the case of read alignment to de Bruijn graphs, the authors prove that their problem formulation is NP-complete. I was skeptical so I set out to see if it had bugs, it didn’t. Then I carefully checked whether it relied on having a large alphabet, nope the proof works with 4 letters. So after much huffing and puffing the proof held and I was convinced. In my review I simply noted
discussion of NP-completeness vs. heuristics: The structure of the DBG, and even
the contig graph is quite specific and not something we would expect to see in practice. The reduction to the Hamilton path also relies on having very long reads, whereas BGREAT is mostly focusing on short reads. Additionally there are several examples where random instances of problems are (provably) solvable using heuristics. I feel that there needs to be a better segue between the NPC result and jumping right into the description of BGREAT.
Any proof of NP-completeness involves showing that the problem you are dealing with now is harder than a problem previously shown to be in NP (and also you should be able to check proofs of it being in NP in polytime). When you do this you take an arbitrary instance of the other problem (in this case a TSP variant) and you encode it in a very special way as a new problem such that if you solve your new problem you can obtain a solution to the old one.
There are two points to make. First the new problem you see (in this case the de Bruijn Graph
The second point I wanted to make is that NP-complete isn’t just about problems being hard, but that the problem is too expressive. This is something I remember Manuel Blum talking about when I first learned NP-completeness properly, I didn’t quite get it at the time, but now I tell myself I do. The issue of expressiveness is useful because it tells us if we are not careful we can accidentally make our problems too hard.
Anyway, that was quite the detour and we haven’t got to the real question posed in the title. Should we really care about NP-complete results in Bioinformatics, i.e. regardless of whether it is true or not (it is in this case), should we let it influence our work, the formulations we propose or new algorithms or whatever.
I’ll argue that the answer is yes, at least in this case. It’s too easy to dismiss this as something from theoryland that has no bearing on reality, just like we routinely solve incredibly large instances of TSP or find cheap flights online (you didn’t click the link above, did you?).
In this specific case the theory to reality disruption happens because 1. the encoding creates a de Bruijn Graph which is quite convoluted and nothing like what we observe in reality, 2. the read constructed in the encoding doesn’t occur in the graph, in particular it doesn’t have to share a single
So we focus on the easier problems, like reads that actually came from the graph or something close to it with not too many errors. Hence the heuristics.
The takeaway is that if we try to solve the general problem, without any restrictions on distance or matching k-mers or anything we cannot guarantee a polynomial time solution unless P=NP.
This is not to say that NP-completeness will always be the right theoretical abstraction to work with. In genome assembly several problems have been proposed as “capturing the essence” of the problem, let’s pick Shortest Common Superstring as our straw man argument. The problem is NP-complete, but it ignores the coverage of repeats when it comes to assembly and so it doesn’t use all the information available. But we cold also ask a different question along the lines of “For this particular genome, how deep do I have to sequence and how long do the reads need to be so that I can solve it with tractable algorithms?”, this is sort of the information theoretic view of assembly, rather than the complexity theoretic view. This is the sort of view that David Tse’s group has been working on.
Thinking about this again I just realized that there is/could be a connection to a previous body of work on graph BWT methods. Notably, the seminal paper by Sirén et al. One of the issues with this approach is that it tends to blow up in memory, when faced with complex variant graphs like chr6 (which contains the infamous MHC region). The advantage of the extended xBWT method is that it doesn’t require to find exact matching k-mers, but the matches can be extended base-by-base just as in regular BWT matching (FM-index to be technically correct). I’ve thought about if there is a way around this issue and my MS student, Tom Schiller, gave it a try, but I don’t think it’s easily solved. Perhaps it is inevitable since it get’s you closer to solving the general problem, which we know is NP-complete.
And this is the real value of NP-completeness for practitioners, it tells you that some problems are so expressive that trying to solve the general case is not going to work.
[Note: there are some issues with the BWT to NP-completeness connection, the extended xBWT requires the graph to be a DAG, I haven’t checked carefully whether the reduction works with a DAG or not. Also even if we had the extended xBWT data structure it’s not necessarily true that we can find an alignment with any cost function on poly-time. This is more like a hunch or an educated guess.]

The key flaw of the paper is that it considers a *path*, not a *walk* (using their own definition). Consider the simple tandem repeat. It would yield the de Bruijn graph with the loop. Consider also error-free read which would span such repeat. How it is possible to even consider *path* to be a reliable solution of the problem?
So, no, it’s not Hamiltonian path vs Eulerian cycle problem, it’s just over-restricted formulation which brings NP-completeness, exactly what you mentioned – “The issue of expressiveness is useful because it tells us if we are not careful we can accidentally make our problems too hard”.
Although the distinction between walk and path is important in the Hamilton Path case (the Hamilton “walk” is just connectivity), once you put a cost function on the path/walk the distinction becomes less important in general.
Whether the proof goes through with the paths relaxed to walks remains to be seen.
The key point about expressivity in this case is that we are willing to consider solutions that are without any seed to start the alignment, i.e. no matching k-mer between the graph and the read, and that the structure of the graph is such that there even if you start at a fixed k-mer (a seed) there are exponentially many paths close to this starting point to consider.
Jouni Siren has a new paper on BWT style indexing of sequence graphs, much more space efficient than their original GCSA, in arxiv, see below. This is being used in more or less bwa style to produce seeds for read alignment in the vg variation graph package (https://github.com/vgteam/vg#vg). It is based on graph transformations and de bruijn graphs, and may be related to your post, and in any case of interest to its readers.
Indexing Variation Graphs
J Sirén – arXiv preprint arXiv:1604.06605, 2016 – arxiv.org
Can’t believe that I missed this, goes to the top of my pile, thanks.
WRT the genome assembly problem, in my opinion the information theoretic view is quite limited in relevance. The work suggests that one should give up whenever the genome is not assemble-able.
In practice for a large fraction of datasets are repeat limited. In this setting, I do not see the point of the coverage depth calculations (which are based on a poisson sampling assumption anyway, which makes their relevance questionable). Isn’t the right question to ask: is given a data-set what is the best assembly that one can get? This leads us back into algorithm design. (of course formally posing that problem can be daunting.)
To the contrary, an information theoretic view of assembly can provide guidance on needed coverage as well as a roadmap to efficient optimal algorithms. See, e.g., http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-S5-S18 and the follow up papers by David Tse and his students/colleagues looking at this problem.
The needed coverage claimed in that paper is completely non predictive. Coverage on real data, and the poisson coverage have almost no correlation. The coverage calculations there serve nothing more than people doing pointless math.
The basic coverage bound of Lander and Waterman is based on uniform iid, or Poisson sampling. There are two dimensions to this assumption, the uniformity and independence. The independence, i.e. that the probability that a specific locus is sequenced is independent of other reads. In HTS this is mostly violated via PCR artifacts and is mitigated with PCR-free library prep, on the whole this is a relatively mild assumption. The uniformity has been shown to be wrong in practice, GC bias for instance affects the probability.
Since those assumptions are violated it is tempting to dismiss this as pointless math.
“All models are wrong, but some are useful” (paraphrasing G.E. Box) applies here, in this case if we assume independent reads the LW bound for the coverage necessary to observe all nucleotides in the genome becomes a lower bound. In the sense that if you vary the probability distribution the minimum coverage required is the one given by the LW.
In the previous comment you mentioned that assemblies were repeat limited, which is precisely the point of the paper linked here. If you consider Figure 2 http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-S5-S18#Fig2 you’ll see that they are considering not just coverage but the length of the reads necessary to beat the repeat regions. And not only lower bounds, but they show the performance of a few algorithms and notably the Multibridging algorithm can almost match the lower bounds presented.
Even in this model they assume that the reads are error free (they are not) and pretty long (2.5Kb). Both of these issues were considered in follow-up papers where the error rate is taken into account and similar results were obtained for paired reads.
I fail to see how this is pointless math.
Let me elaborate on why I think this paper (http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-S5-S18) is pointless math (for a practitioners like me). There are two main points which lead me to question the point of research like this:
1. The authors claim that there are some fundamental read lengths needed to assembly. This depends upon the genome being assembled. If we knew the genome being assembled, there would be no point to assembly :). If the paper had at least talked about estimating the read length needed to assemble from data at least, I’d retract my claim about its pointlessness.
2. The authors seem to restrict themselves to the case when assembly to one contig is possible. The authors seem to be claiming that the their algorithm is “optimal” because it can reconstruct sequences when they can be completely reconstructed from data. This is a laughable notion of optimality. As a consumer of assembly software in a job that has often required me to get assembly of organisms from high throughput data for the last few years years, this is a very rare event (especially for short reads, and anything beyond small bacteria from long reads). The question that I’d want the theorists to answer would be “What should I do to get the most out of the data I already have?.” This approach does not answer that question at all.
LW is a relevant statistic as I can get an estimate of the genome length and know how much I should I must sequence to have any hope of getting it. Of course it’s not exact, but captures some intuition. The calculations of this paper not only assume a uniform sampling model, but also are fairly useless to me as they are genome dependent.